“衰老”我国科学家揭示逆转心脏衰老的关键蛋白
今天,很高兴为大家分享来自媒体滚动的我国科学家揭示逆转心脏衰老的关键蛋白,如果您对我国科学家揭示逆转心脏衰老的关键蛋白感兴趣,请往下看。
衰老是心血管疾病的首要危险因素,可导致心脏结构异常和功能衰退,如室壁肥厚、舒张功能障碍、纤维性颤动等。这些年龄相关的心脏变化往往会增加多种心脏疾病的患病率,进而影响人类健康和寿命。随着全球人口老龄化形势日益严峻,探索人类心脏衰老的核心机制,制定相应的预警、预防和治疗策略变得尤为重要。
心脏衰老是一个复杂的动态过程,受到多种因素的影响。迄今为止,关于灵长类心脏衰老的跨维度研究仍鲜有报道,其关键分子机制亟待揭示。
2023年10月3日,中国科学院动物研究所刘光慧课题组、曲静课题组和中国科学院北京基因组研究所张维绮课题组合作在Nature Aging杂志在线发表题为“SIRT2 counteracts primate cardiac aging via deacetylation of STAT3 that silences CDKN2B“的研究论文。该研究首次基于食蟹猴衰老心脏转录组和蛋白组的联合分析,揭示SIRT2通过去乙酰化STAT3进而延缓灵长类心肌衰老的新机制,并通过SIRT2基因疗法实现了衰老心脏功能的逆转。
该研究入选Nature Aging当期封面文章,杂志同期配发了News & Views 对论文进行了亮点评述。
在这项工作中,研究人员利用自然衰老的非人灵长类动物(食蟹猴)模型,通过对心脏肥大、肌节紊乱和炎症等衰老表型分析,结合高通量蛋白组和转录组测序、基因编辑、人多能干细胞定向诱导心肌细胞分化、基因表达调控、病毒载体基因治疗等技术手段,发现SIRT2蛋白的表达下调是驱动灵长类心肌细胞衰老的关键因素。一方面,研究人员通过对食蟹猴心脏中鉴定到的衰老差异蛋白与不同蛋白编码基因集(包括衰老相关的心血管疾病相关基因、Aging Atlas衰老相关基因及表观调控相关基因)进行联合分析,发现去乙酰化酶SIRT2是唯一与不同心血管疾病以及表观遗传调控均相关的衰老下调蛋白。另一方面,研究人员利用CRISPR-Cas9介导的基因编辑和人多能干细胞定向诱导分化技术,首次获得了SIRT2敲除的人心肌细胞,并发现SIRT2缺失的人心肌细胞表现出加速衰老、异常肥大等一系列心脏衰老相关表型,且表现出与老年食蟹猴心脏类似的基因表达谱改变。
接下来,研究人员结合转录因子网络解析,免疫共沉淀偶联蛋白质谱分析、蛋白定点突变、染色质免疫沉淀-实时定量聚合酶链反应、原位电镜等多种技术方法进行了深入的机制研究,发现SIRT2蛋白通过与转录因子STAT3相互作用,促使后者K685去乙酰化,进而抑制细胞周期阻滞基因CDKN2B的转录。SIRT2的缺乏引起人心肌细胞中STAT3的乙酰化水平升高,促进CDKN2B基因表达,导致心肌细胞衰老和肥大。与之相对,过表达SIRT2或敲低CDKN2B均可延缓人心肌细胞衰老。
在证明SIRT2是调控人心肌衰老的关键靶标后,研究人员提出基于SIRT2的基因疗法或许可延缓心脏衰老这一科学假设。为了测试这一点,研究人员向24月龄老年小鼠心肌内多点注射编码SIRT2蛋白的慢病毒,并在2周后发现老年小鼠心脏的射血分数(EF)、短轴缩短率(FS)以及心肌肥大等指标均表现为明显的“年轻化”特征,提示SIRT2可作为体内干预心肌衰老的分子开关,今后或可通过发展SIRT2增强基因疗法或SIRT2特异性激动剂实现对心脏衰老及相关心血管疾病的预防和治疗。
综上,该研究系统绘制了灵长类心脏衰老的多维基因表达图谱,深入解析了SIRT2-STAT3-CDKN2B通路在灵长类心肌细胞衰老中的关键作用,揭示了可逆转心脏衰老的“乙酰化开关”。该研究为明确人类心脏衰老的分子驱动力、建立心脏衰老及相关疾病的临床诊断、早期预警及干预策略提供了重要的线索和思路。
该研究由中国科学院动物研究所、中国科学院北京基因组研究所、北京干细胞与再生医学研究院、首都医科大学宣武医院等多家机构合作完成。中国科学院动物研究所叶燕霞助理研究员、中国科学院北京基因组研究所硕士生杨宽、湖南大学刘海松教授、北京大学第三医院于洋研究员以及中国科学院动物研究所宋默识研究员为并列第一作者。中国科学院动物研究所刘光慧研究员、曲静研究员及中国科学院北京基因组研究所张维绮研究员为文章的共同通讯作者。该研究获得科技部、国家自然科学基金委、中国科学院和北京市等项目资助。
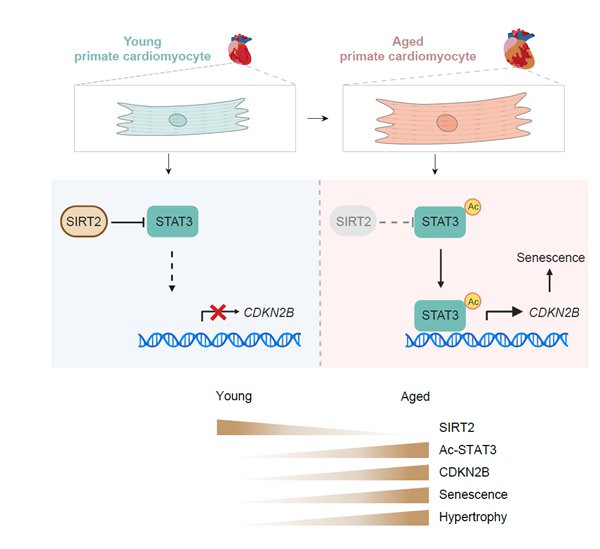 图1. SIRT2延缓灵长类心肌衰老的分子机制
图1. SIRT2延缓灵长类心肌衰老的分子机制 图2. 利用SIRT2编织年轻健康心脏(入选Nature Aging杂志封面)
图2. 利用SIRT2编织年轻健康心脏(入选Nature Aging杂志封面)(原标题:动物研究所合作揭示逆转心脏衰老的关键蛋白)
好了,关于我国科学家揭示逆转心脏衰老的关键蛋白就讲到这。
版权及免责声明:凡本网所属版权作品,转载时须获得授权并注明来源“科技金融网”,违者本网将保留追究其相关法律责任的权力。凡转载文章,不代表本网观点和立场,如有侵权,请联系我们删除。
相关文章
- “在我”忆来只把旧书读
- “科博会”芜湖科博会集中展示大国重器
- “诗人”带着大海散步的人
- “宁德”宁德时代三季报:业绩增速放缓、海外市场份额扩大
- “新材料”信金控股完成新一期人民币基金首关
- “同比增长”失守3000点后,A股市场的危与机|智氪
- “商务部”商务部:达成共识!
- “基金”又一家基金公司换董事长,年内公募高管变动人数增至321人
- “模型”解决大模型复现难、协作难, 这支95后学生团队打造了一个国产AI开源社区
- “教师”北京化工大学回应学院院长被指骚扰教师:已成立工作专班,对师德失范问题零容忍
- “鱼类”已知最原始的铰齿鱼“现身”
- “小鼠”35岁博士一项成果磨6年:科研中“试”的过程非常熬人
- “转录”中国科学院揭示热纤梭菌转录调控因子σI的独特启动子识别机制
- “空间科学”第三届中国空间科学大会开幕
- “衰老”中国科学家揭示人类基因组中的“年轻化”基因
- “植物学”3人获奖,第四届吴征镒植物学奖获奖名单公示
- “中国科学院”王友绍当选2023国际先进材料协会会士
- “神经”科学家揭示人脑中注意对神经活动共变性的调节机制
- “奋斗者”他用“小微球”助力“奋斗者”号突破万米深潜
- “中心”中国科学院成都分院与国家空间科学中心共建π中心









