“催化剂”打破催化剂设计“天花板”,科学家提出图神经网络模型,助力实现材料性能的跨尺度模拟
今天,很高兴为大家分享来自DeepTech深科技的打破催化剂设计“天花板”,科学家提出图神经网络模型,助力实现材料性能的跨尺度模拟,如果您对打破催化剂设计“天花板”,科学家提出图神经网络模型,助力实现材料性能的跨尺度模拟感兴趣,请往下看。
来源:DeepTech深科技
“我们开发了一款机器学习模型,并将其用于高熵合金这一新材料中。测试结果显示,AGAT 模型的预测速度比第一性原理计算速度快 4 万倍。而且这款模型是通用的,能满足不同场景之下的先进材料设计。我们还公布了源代码,以帮助读者重复论文结果和推动实际应用。”香港城市大学教授赵仕俊表示。


预测速度提高 40000 倍
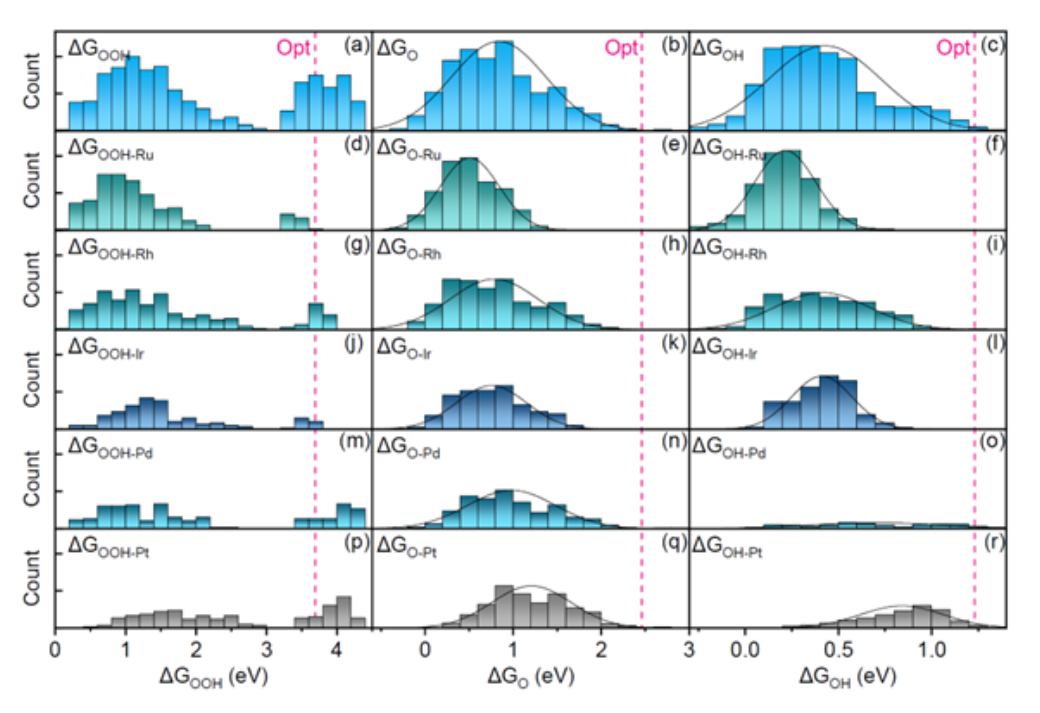
据介绍,这款机器学习模型的名字叫做图神经网络模型(AGAT,atomic graph attention)。该模型可被用于优化吸附质的吸附构型,借此可以获得反应中间体在高熵催化剂表面的吸附能分布。
赵仕俊说:“大部分深度学习模型是黑箱模型,人类无法或很难理解模型的预测逻辑。而 AGAT 模型的关键可训练参数是可以被解释的,通过这些参数可以有效理解 AGAT 模型的预测行为。”
通过产生大批量的数据,高熵电催化剂可以凭借丰富的表面吸附位点,来绕过线性关系的限制。同时,AGAT 模型可以发现并预测高催化活性组分。
另外,研究中该团队成功制备了多个高熵合金催化剂,并发现它们的催化活性高于铂催化剂。
由于 AGAT 模型可以预测原子的受力与能量,故可作为深度学习势能来使用,并在以下三个方面具备广阔的应用前景:
其一,AGAT 模型可用于结构材料形变的大尺度模拟。不同的材料模拟方法的特点各不相同,因此往往用于不同的时间尺度与空间尺度。
第一性原理有着精度高、计算速度慢的特点,因此更多用于低于 1000 个原子的纳米体系与亚纳米体系的计算与模拟,对于较为复杂的位错和晶界等缺陷,第一性原理的模拟往往显得力不从心。
基于经验势的分子动力学方法,固然可以模拟较大的体系。但是,其精度低于第一性原理。而使用 AGAT 模型训练出来的势能,具备第一性原理的精度,且计算速度远快于第一性原理。因此,AGAT 模型可用于模拟较大的体系,从而拓宽高精度模拟的时间尺度与空间尺度。
其二,AGAT 模型可用于辅助新型药物的开发。由于它不仅可以模拟有周期性边界条件的固体体系,还能用来模拟小分子,故可以协助新型药物的开发与模拟。
其三,AGAT 模型可用于模拟复杂催化界面与电池电极界面。在多相催化和电池电极的真实环境中,界面反应可谓极其复杂。受到第一性原理时间尺度与空间尺度的限制,采用传统方法往往只能计算单个或少量吸附质在表面的吸附,很难模拟反应物在液固或气液固条件之下的反应过程。而 AGAT 模型拥有较高的精度和计算速度,故能模拟这种复杂的化学反应。
 (来源:Joule)
(来源:Joule)
如何绕过提示催化剂效果的“天花板”?
尽管这是一项“AI+Science”的新颖研究,但其背后却蕴含着一个宏大命题。能源危机与环境污染,是当今世界面临的主要问题,且与经济民生息息相关。
电催化,是清洁能源转化过程中的重要一环。在电催化的过程中,催化剂扮演着关键角色。
而电催化剂的优化与设计,是提高清洁能源的转化效率时必须克服的重要短板之一。传统催化剂比如二氧化铱、二氧化钌和铂,由于具有较好的催化效率,已被广泛用于电催化反应。
然而,这些催化剂的成本非常高昂,特别是铱、钌和铂的价格,几乎和黄金价格相当。尽管人们已经采用诸多方法来对其进行优化,比如修改催化剂的结构和形貌、调整催化剂与载体的相互作用、精细调整活性中心的局域环境等。
但是,对于提升催化剂效果来说,目前这些方法已经触及“天花板”,即始终无法逾越催化理论中的线性关系。所以,若想进一步提升催化剂的性能,必须使用新的设计理念和新的催化剂类型。
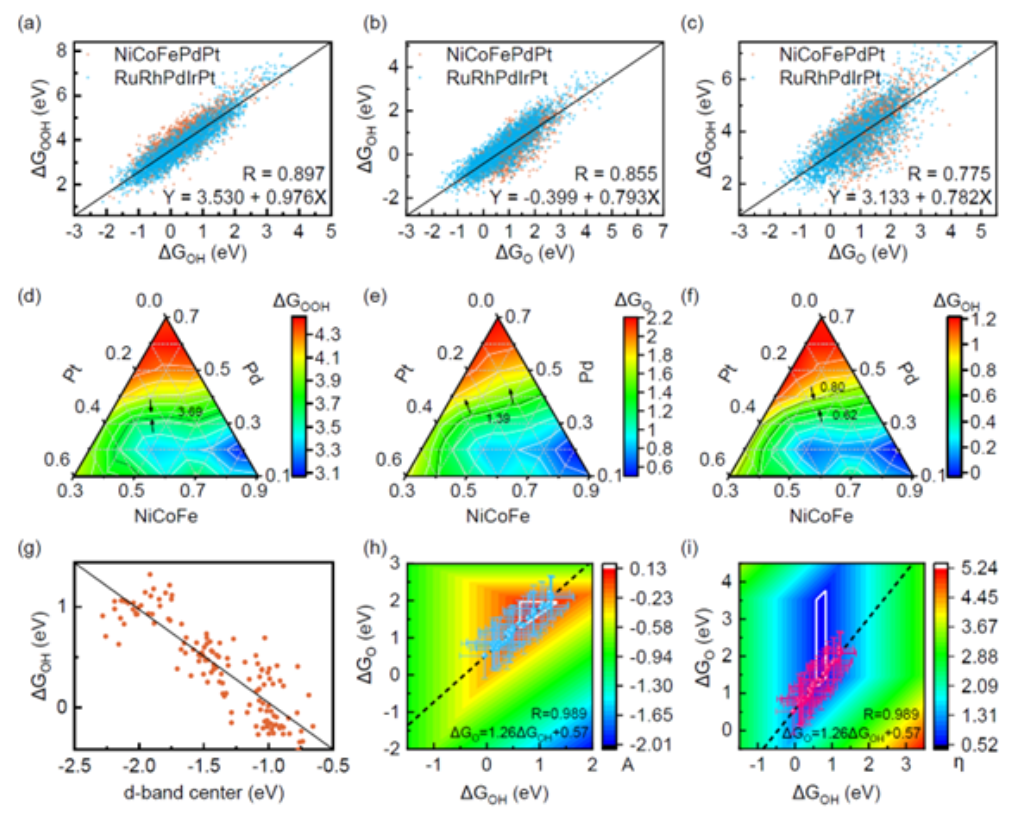
在本次研究中,课题组通过引进最新的材料设计理念——高熵合金,来提高传统催化剂的性能、以及降低催化剂的成本。
 (来源:Joule)
(来源:Joule)高熵合金,是一类含有五种或更多主要元素的无序合金。在这种合金中,不同金属元素呈现出随机或接近随机的状态分布,其体系的混乱程度较大、构型熵也比较高。
21 世纪初以来,高熵合金的应用早已“遍地开花”,人们将其用作结构材料、防腐蚀材料、抗辐照材料等。在高熵合金材料中,由于存在多种元素,所以其表面的活性位点非常丰富,相组成空间也比较广阔,并且高熵合金还具备出色的动态稳定性,这为设计高效催化剂提供了很好的平台。
另外,高熵合金还具有独特的化学无序性和局域环境的畸变,故其有潜力去打破线性关系的约束。
近年来,一些先进的合成方法已被用于合成高熵合金纳米团簇。在高熵合金的帮助之下,甚至那些传统认知中互不相溶的组分也能被合成出来,这让高熵合金俨然成为了催化领域的“香饽饽”。
然而,由于高熵合金的组分空间巨大,而且其表面位点较多,因此在设计以及优化高熵催化剂的组成和位点时,依然面临着巨大的挑战。
例如,为了获得吸附质在某一组分表面的吸附性质,必须进行大量的计算。当使用计算模拟的方法来制备高熵催化剂时,相应的计算成本始终居高不下。
基于上述考量,赵仕俊和团队想到了机器学习的方法。
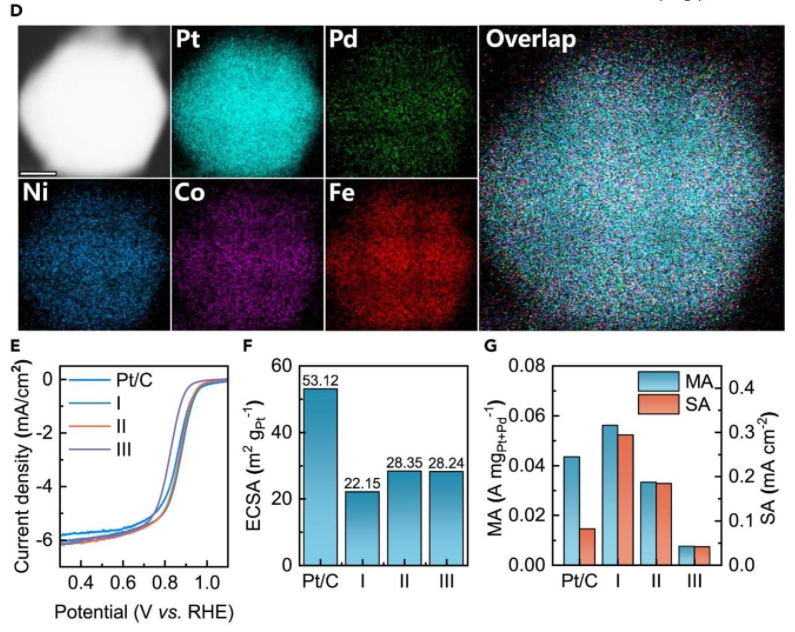
近年来,将机器学习用于设计高熵材料早已不是新鲜事。然而,传统机器学习模型的预测精度较低,这导致其应用场景较为局限。而且,在传统机器学习模型描述符的选取上,高度依赖于研究者自身的专业知识。
而对于催化剂设计来说,最为关键的物理参量是吸附质的吸附自由能。这一物理量与局域原子的排布息息相关,因此必须考虑局域原子排布的影响。
另外,现有的图神经网络模型大多基于图卷积神经网络,而图卷积神经网络中的原子以相同的权重,汇聚了周围所有的原子信息,也就是说近邻原子相互之间的影响是一样的。
但实际上,对于距离较远的原子对来说,它们之间的相互影响程度较低,故无法与图卷积神经网络的消息传递机制保持一致。
在催化过程中,由于涉及到不同吸附质的吸附性质,因此预计这一吸附性质主要由距离最近的原子来决定。
基于此,该团队决定改进传统的图神经网络,即引入不同原子排布之间的不同贡献、以及各种对称不变性,这让 AGAT 模型可以有效规避复杂的特征工程,从而降低模型对于研究者专业经验的依赖,并能在预测晶体与分子的性质时展现出一定优势。
 (来源:Joule)
(来源:Joule)在性能上,他们计划进一步优化模型结构,以实现更高的训练精度和训练速度;在应用上,其打算将 AGAT 模型融入主动学习流程中,使其可以自动训练并能发现更好的催化剂成分,从而降低模型对研究者经验的依赖。另外,过渡态搜索和分子动力学模拟这两个功能,也将被纳入 AGAT 模型之中。
 参考资料:
参考资料:1.Zhang, J., Wang, C., Huang, S., Xiang, X., Xiong, Y., Xu, B., ... & Zhao, S. (2023). Design high-entropy electrocatalyst via interpretable deep graph attention learning.Joule.
排版:朵克斯
好了,关于打破催化剂设计“天花板”,科学家提出图神经网络模型,助力实现材料性能的跨尺度模拟就讲到这。
版权及免责声明:凡本网所属版权作品,转载时须获得授权并注明来源“科技金融网”,违者本网将保留追究其相关法律责任的权力。凡转载文章,不代表本网观点和立场,如有侵权,请联系我们删除。
相关文章
- “铜陵”化工厂爆炸?两名造谣者被查处
- “爷爷”爷爷的石榴树
- “陈老”高山仰止,景行行止 ——读春桃老师所著《国医》
- “知网”中国知网用户委员会两名成员公开亮相
- “宜宾”近2万人参加!2023宜宾长江马拉松开跑:埃塞俄比亚选手包揽全马组男女前三名
- “鲁南”鲁南制药集团建厂55周年:“向新向未来”
- “民谣”玉林民谣,从成都走向深圳
- “亿元”爱仕达董事长陈合林做铁锅起家 公司已连续亏损两年多他有啥招术?
- “营收”“酱油一哥”黯然失色!市值蒸发超5000亿,海天味业遭转型阵痛
- “可持续”“京澳25”公益计划启航
- “模型”解决大模型复现难、协作难, 这支95后学生团队打造了一个国产AI开源社区
- “模型”人工智能公司OpenCSG发布大模型开源生态社区“传神”
- “模型”北理工团队在人工智能图像识别领域取得新进展
- “模型”参数少近一半,性能逼近谷歌Minerva,又一个数学大模型开源了
- “模型”评论能力强于GPT-4,上交开源13B评估大模型Auto-J
- “模型”端侧首次实现70亿AI语言大模型 联发科vivo强强联手
- “模型”中国信通院承接工信部大模型公共服务平台建设工作,联合 360、京东、商汤等
- “融资”国内AI大模型赛道火热,大厂积极跟投布局
- “模型”在RTX 4090被限制的时代下,让大模型使用RLHF更高效的方法来了
- “图像”OpenAI终于Open一回:DALL-E 3论文公布、上线ChatGPT,作者一半是华人









