“分子”KDD 2023 | 第四范式开发用于分子性质预测的生成式3D预训练模型
今天,很高兴为大家分享来自机器之心Pro的KDD 2023 | 第四范式开发用于分子性质预测的生成式3D预训练模型,如果您对KDD 2023 | 第四范式开发用于分子性质预测的生成式3D预训练模型感兴趣,请往下看。
机器之心专栏
作者:4Paradigm AutoGraph团队

论文标题:Automated 3D Pre-Training for Molecular Property Prediction

论文链接:https://arxiv.org/abs/2306.07812
代码链接:https://github.com/LARS-research/3D-PGT
该论文已经被KDD 2023接收。
1. 简介
分子性质预测是药物发现和材料科学等领域中的一个重要问题。考虑到分子的3D结构信息与其性质紧密相关,近年来,一个主流的研究热点是将分子的3D结构信息与各种以图学习为代表的机器学习方法结合,以提高分子性质的预测性能。然而,由于高昂的计算成本,在大规模数据实时计算分子的3D结构几乎是不可行的[1]。
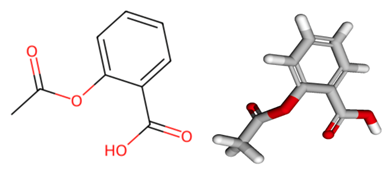 图1:cc(=o)oc1=cc=cc=c1c(=o)o的2D分子图(左)和3D构象图(右)。
图1:cc(=o)oc1=cc=cc=c1c(=o)o的2D分子图(左)和3D构象图(右)。面对这一挑战,我们提出了一个以预训练范式为基础的大模型,专门用于分子性质预测。预训练范式隶属于 AIGC 的一个重要技术分支[2],其能处理大规模数据集并提取深层次特征,充分利用了大模型的优势。本文在分子领域结合已有的3D分子构象进行预训练,并在实际下游任务中只基于分子的2D结构信息进行微调并进行性质预测,从而在保证高效的前提下进一步提升在下游任务上的表现。
在这项工作中,我们提出了一种自动化3D预训练框架:3D-PGT。基于分子的化学键长、键角和二面角是对应于完整分子3D构象的三个基本几何描述符这一事实,我们对应设计了三个生成式预训练任务,使得模型通过预训练能够具备编码3D几何结构的能力。而为了自动化分配这三个预训练任务的权重以融合成一个总体的预训练目标函数,我们基于分子总能量设计了一个surrogate metric,从而自动搜索三个预训练任务的权重分布。
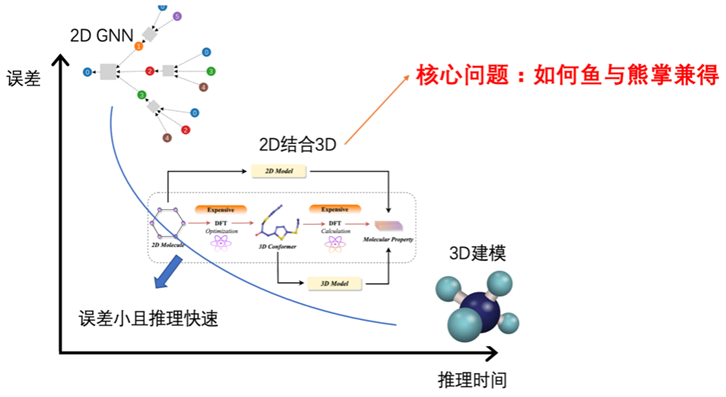 图2:分子性质预测问题的核心问题示意:如何兼具更快的推理速度和更小的推理误差。
图2:分子性质预测问题的核心问题示意:如何兼具更快的推理速度和更小的推理误差。为了验证所设计的预训练框架的有效性,我们基于已测定 3D 结构的公开分子数据集进行了预训练,并在 8 个量子化学性质预测任务和 12 个涉及药理学、分子化学的下游任务上进行微调和实验验证。结果表明,3D-PGT能通过3D预训练带来明显的性能增益,且优于其他预训练 baseline。这再次证明了在分子性质预测领域,大模型处理复杂任务和大规模数据的优势。同时,基于 3D-PGT 的方法还在催化剂仿真挑战 Open Catalyst Challenge 2022 上取得第 3,在大规模分子性质预测挑战 OGB-LSC@NeruIPS challenge 2022 中取得第 8 名的成绩。
2. 背景介绍
分子性质作为描述分子特征和行为的基本属性,在药理学、材料化学等研究领域和具体应用都有着重要意义。然而分子性质的测定往往需要借助成本高昂的计算方式,例如以密度泛函理论(Density Functional Theory, DFT)为代表的计算化学方法。近年来,随着大规模量子化学计算和高通量实验的技术进步,一个新兴的工业界和学术界热门的研究方向,是利用具有适当归纳偏置的机器学习方法并结合已测定性质的大量分子数据,实现高效的分子性质预测,并应用于一系列实际下游应用中,例如大规模药物分子筛选,合成材料筛选,催化剂设计等。

图3:分子性质预测效率对比示意图。其中基于量子化学计算的DFT计算特定分子性质需要数个小时,而利用机器学习方法只需要远小于1秒的时间。该图出自OGB@NeurIPS 2022 Challenge,预测分子的HOMO-LUMO能隙。
在过去的几年中,一种主流的方法是将分子建模为 2D 图结构,其中以原子作为节点,而边作为化学键,将整个分子性质预测建模为图级(Graph-level)预测任务,并应用 GNN(Graph Neural Network)通过拟合 DFT 的计算方式预测分子的性质。但目前,这种方法只有较高的预测效率,预测性能距离实际应用还有较大的差距。

图4:现有分子性质预测方法的一般流程。基于DFT的量子化学计算方法首先对分子的三维结构进行优化以获得低能构象,然后基于构象计算特定的分子性质。2D模型近似DFT的整个过程,通过输入的2D分子图直接预测分子性质;3D模型在2D分子图的基础上还需要进一步输入3D分子构象,以获得更好的预测性能。
由于分子的 3D 结构反映了原子和官能团之间的相互作用和相对位置,因此 3D 结构对分子性质的理解至关重要。考虑到这一点,一系列方法针对分子的 3D 结构信息设计 3D 模型,从而获得更好的预测效果。然而,3D 结构的获取需要依赖 DFT 等量子化学计算手段,这种昂贵的计算成本导致分子的 3D 结构在许多实际的下游任务中往往是不可获取的。
结合上述两种主流路线各自的优缺点,一个最近比较新颖的角度是将包含已测定 3D 结构信息的数据集作为预训练数据集,基于 3D 结构设计预训练任务,从而让模型理解 DFT 由 2D 分子图计算优化 3D 结构的过程,并将该先验信息迁移到下游 2D 分子性质预测任务上,从而兼具效率和性能。目前,主流的同期工作例如 GraphMVP[3] 和3D Infomax[4] 都通过对齐分子 2D 视图和 3D 视图的图级表示向量来设计预训练目标函数,并通过在主流 benchmark 上的实验证明了 3D 预训练方案的有效性。
综上,本文继续沿着分子 3D-Pretraining 的 Pipeline,设计了三个基础的生成式预训练任务,并基于分子总能量设计目标函数来自动搜索各预训练任务的权重,从而构建多预训练任务的自动融合框架,并在广泛的下游任务上获得了显著的预训练收益。
3. 本文的方法
3.1 生成式预训练任务的设计
DFT 优化分子 3D 结构的核心是在势能面上寻找局部最小值[5],分子能量出于势能面上局部最小值时的 3D 结构也被称为分子构象。其中,构象所包含的 3D 几何信息可以被以下三个描述符完整地描述:两个原子之间的化学键长、两个化学键之间形成的夹角,以及三个化学键形成的二面角。
生成式预训练任务使模型能够理解分子从 2D 拓扑到 3D 几何的基于 DFT 的优化过程。由于分子几何是由电子的量子力学行为决定的,因此生成式预训练任务可以通过学习 3D 构象的生成来间接学习量子化学性质的预测。基于此,本文设计了三个生成式预训练任务来分别生成这键长、键角、二面角这三个描述符,具体如下所示:

图5:分子结构和势能面关系示意图。势能面将分子能量定义为多个坐标轴的函数,分子3D构象的优化就是在势能面上寻找局部最小值,而3D构象中的几何信息可以通过连接两个原子的键长、三个相连原子的键角和三个连续键的二面角来描述
其中,h 代表对应节点的表示向量,f 为对应的预测网络(这里的设置是MLP)。可以看到,这三个任务的核心思路都是利用 backbone 提取的节点表示来设计具体的回归任务,通过将描述符中包含的 3D 结构信息当做解读信号,从而让 backbone 具有编码 3D 结构信息的能力,并将这种能力作为先验嵌入到模型并迁移到实际的下游任务中。
同时,考虑到当邻居数为|N|时,分子的键角和二面角的计算复杂度呈 Ο(|N|^2 )和Ο(|N|^3 ) 增长,使得大规模的 3D 预训练实现成本过高。本文基于 RGC(Runtime Geometry Calculation)重新设计了键角和二面角的目标函数,用每个原子的所涉及的键角和与每个化学键设计的二面角的和来代替所有键角和二面角的预测,从而将计算复杂度降低到线性级别。
3.2 自动化多预训练任务融合框架
由于我们同时定义了多个预训练任务,且各预训练任务的损失函数对于模型参数的梯度优化方向并不一致,因此各自预训练任务的权重分配是一个需要考虑的问题。如何自动化且最更好地分配多预训练任务的权重,以预期在下游任务上获得更好的性能增益,是设计多预训练任务融合框架的核心。
由于键长、键角和二面角都是局部描述符,因此这里需要一个明确的 surrogate metric 来评估预训练后的 backbone 对于分子 3D 结构整体的编码能力而不是局部编码能力。对此,本文考虑得到分子总能量和分子 3D 结构之间的对应关系,基于分子总能量设计了一个目标函数,并基于此设计了 bi-level 的油画框架来搜索三个生成式预训练任务各自的权重。最终,我们整体预训练的 pipeline 如下图所示。在预训练阶段,我们首先基于预训练分子的低能构象设计了三个生成式预训练任务,并基于分子低能构象对应的分子总能量设计了一个 surrogate metric 来搜索三个预训练任务的各自权重;而在下游的微调阶段,由于我们已经在预训练阶段引入了分子几何先验,因此通过微调即可在实际下游任务中获得性能增益。
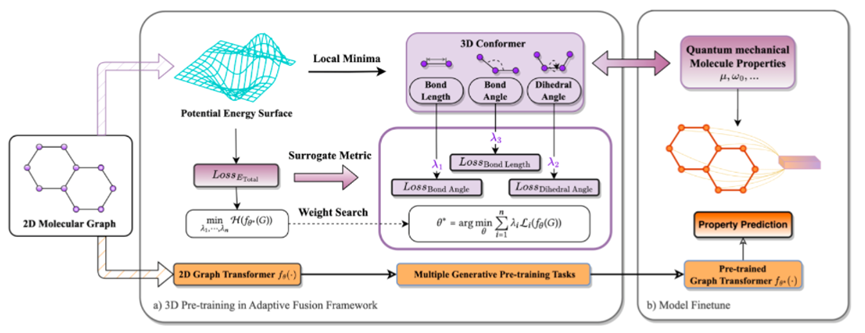 图6:3D-PGT的预训练框架示意图
图6:3D-PGT的预训练框架示意图4. 实验结果
在本文中,我们针对性的设计了一系列实验来证明 3D 预训练这一技术路线和本文设计的预训练任务及自动融合框架的有效性,从而面临当只有 2D 分子图可以用于预测时,可以避免为每个分子生成 3D 构象的巨大计算成本,并能通过微调获得明显的性能收益。
本文主要在 3 个包含 3D 结构信息的数据集上进行预训练,并主要在 12 个下游任务上进行微调和效果验证,数据集的详细统计数据如下所示:
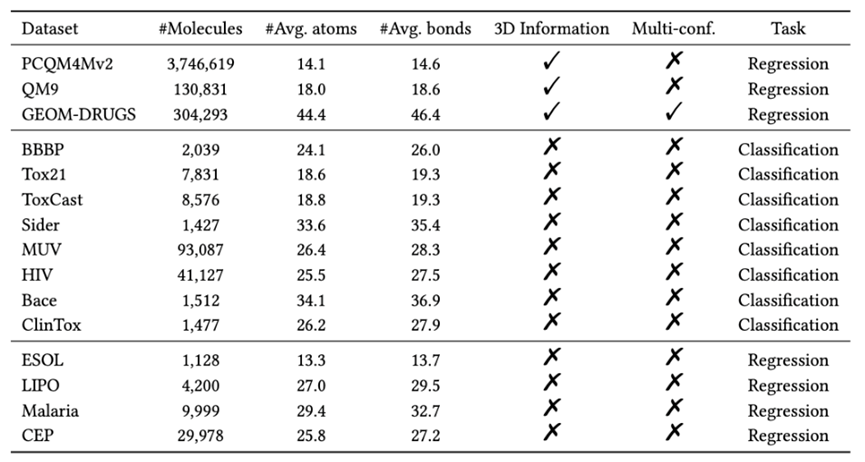 图7:数据集的统计细节
图7:数据集的统计细节4.1 量子化学性质预测的表现
本文首先在流行的基准数据集QM9上评估了3D-PGT的量子化学领域的性质预测能力。我们首先从QM9包含的134k单一分子构象样本中随机选取50k个携带构象信息的分子样本用于预训练,并从剩余的样本中选取50k个分子样本并屏蔽其3D结构信息用于微调和评测。结果如下:
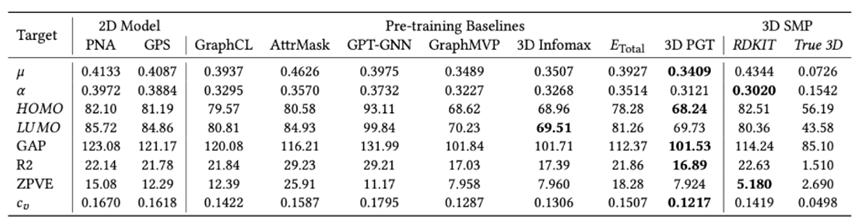 图8:QM9数据集实验对比结果,评价指标为MAE(平均绝对误差)
图8:QM9数据集实验对比结果,评价指标为MAE(平均绝对误差)从实验结果中,我们观测到以下现象 :1)首先,3D-PGT以GPS作为backbone进行预训练和微调,但相较于GPS的baseline,3D-PGT在8种量子化学性质的预测上平均减少了17.7%的MAE;2)同时,3D-PGT也明显优于其他2D预训练方法,以GraphCL为例,其不包含3D结构信息的预训练所能带来的性能提升是有限的;3)相较于GraphMVP和3D Infomax两种前沿的3D预训练方法,我们的预训练框架仍然具有性能优势;4)值得注意的是,当给定准确的3D结构时,3D模型SMP取得了显著的性能优势,但当提供给SMP的3D信息来源是粗糙的RDKit计算结果时,3D-PGT在8个预测任务中的6个都击败了SMP,体现了3D预训练范式在效率和性能上优势。
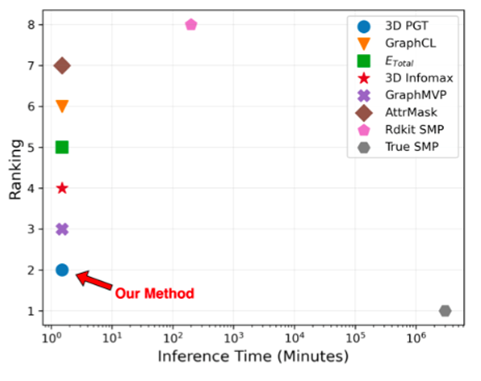 图9:不同方法在HOMO预测任务上的推理时间和性能排名
图9:不同方法在HOMO预测任务上的推理时间和性能排名4.2 在其他广泛下游任务上的泛化能力
除了预测与分子 3D 结构密切相关的量子化学性质外,我们还将下游任务进一步推广到药理学、物理学和生物学等领域,且这些任务的输入只包含 2D 分子图。我们在 GEOM 数据集中随机选择 50 k个具有单个构象的分子进行预训练,并对 8 个主流下游分子性质分类任务和 6 个回归任务上进行微调,且这些下游任务包含的数据量都较少,具体结果如下:
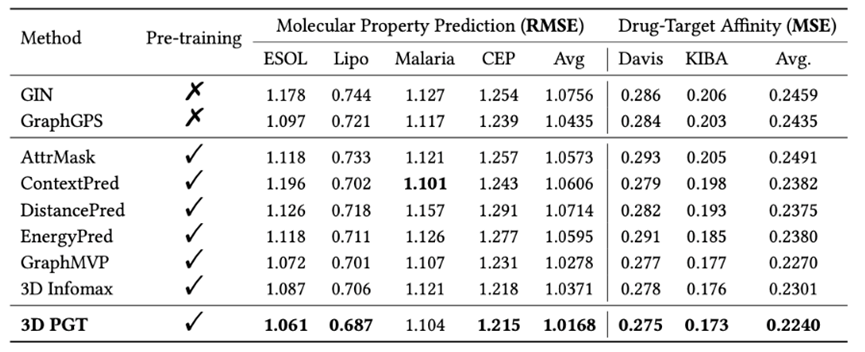 图10:在四个分子性质预测任务(RMSE)和两个药物蛋白靶点亲和力预测任务(MSE)上的实验结果
图10:在四个分子性质预测任务(RMSE)和两个药物蛋白靶点亲和力预测任务(MSE)上的实验结果可以看到,3D-PGT 在大多数下游任务上都优于其他 baseline,且与 GPS 相比,3D-PGT 依旧可以预训练获得稳定的性能收益。这意味着我们所设计的预训练框架可以将性能增益推广到广泛的下游任务上(除分子性质预测本身外,在两个药物和靶蛋白亲和力预测任务 Davis 和 KIBA 上也获得了性能收益),而不局限于和 3D 结构密切相关的量子化学性质预测,且即使在小样本数据上的微调也能带来性能增益。
4.3 在大规模数据挑战赛上的表现
在 OGB-LSC 的 Graph-level 预测任务赛道中,PCQM4Mv2 是一个包含 374 万分子的大型分子数据集,其中的 337 万个训练样本的 3D 几何信息通过 DFT 计算得到,且为了接近大规模的虚拟筛选场景,该挑战没有提供验证集和测试集的 3D 构象,且要求使用单个 GPU 在 4 小时内完成 150k 个分子的 HOMO-LUMO gap 的推断,这意味着在模型的推理阶段计算所有测试样本的几何结构是不可行的。3D-PGT 在该数据集上的表现与 Leaderboard 排名前列的方法对比结果如下所示:
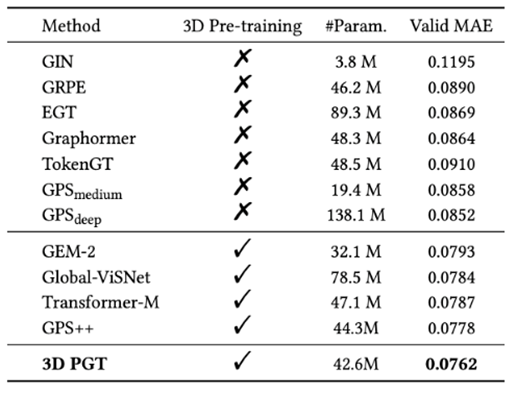 图11:在PCQM4Mv@验证集上的实验结果
图11:在PCQM4Mv@验证集上的实验结果可以看到,与现有的不考虑 3D 结构信息的 GNN 和 Transformer 系列方法相比,3D-PGT 通过引入生成式预训练任务得到了明显的性能提升。其中针对GPS 的 baseline,3D-PGT 通过设计的自动预训练框架降低了 10.6 的 MAE,且在单模型的性能上由于OGB@NeruIPS 2022冠军方案GPS++。
与此同时,Open Catalyst Challenge 2022是一个致力于利用分子模拟发现高性能催化剂的挑战赛。该比赛的核心任务是之一利用机器学习模型预测催化剂分子活性。训练集包含 200 万个分子,且记录了每个分子样本的 3D 结构和总能量。最终,结合 AutoGraph 的算法,3D-PGT 在该挑战赛上取得了第三的成绩,再次印证了该预训练框架的有效性和竞争力。
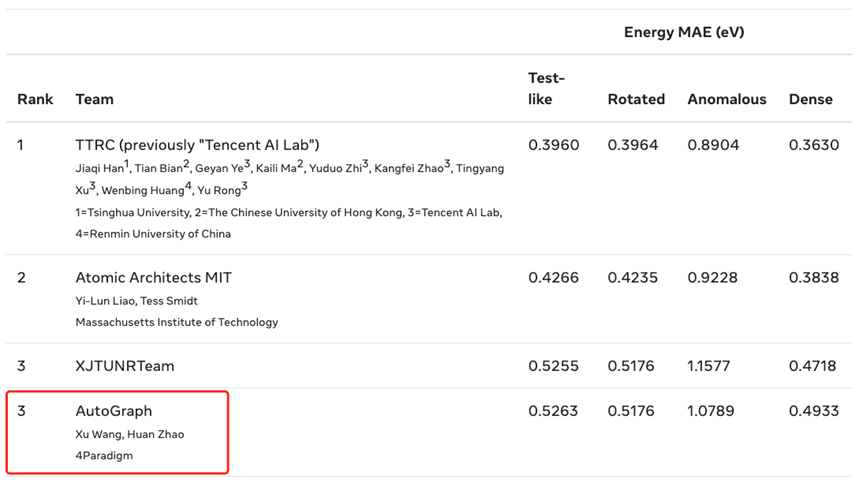 图12:Open Catalyst Challenge 2022 Leaderboard结果
图12:Open Catalyst Challenge 2022 Leaderboard结果5. 结论
在这项工作中,我们提出了 3D-PGT,一种自动化的 3D 预训练框架,专注于提升在实 3D 结构不可用的分子性质预测任务上的表现。
3D-PGT 设计了多个生成式预训练任务,这些任务可以将几何先验带入微调阶段。同时,为了更好地自动融合这些预训练任务并使其效益能够具有普适性,我们基于分子总能量设计了一个预训练的 surrogate metric 来搜索每个预任务的自适应权重。本文基于现有主流的量子化学数据集和一系列广泛的下游任务设计了实验,且实验结果表明,3D-PGT 通过预训练引入潜在的几何先验不仅有利于量子化学性质的预测,而且有利于药理学、物理化学和生物物理学等领域的预测。此外,在 OGB 排行榜上,3D-PGT 在大规模分子预测方面优于顶级解决方案的所有 baseline。
在未来的工作中,考虑到在 NLP 领域 GPT 的预训练范式的成功,我们会将该框架和 3D 分子预训练范式推广到实际的下游应用中,例如开发用于储存可再生能源的催化剂等。
注:该论文已被KDD 2023接收,如有问题,请联系[email protected]。
参考文献
[1] Learning neural generative dynamics for molecular conformation generation. Arxiv 2021
[2] Language models are few-shot learners. NeurIPS 2020
[3] Pre-training Molecular Graph Representation with 3D Geometry. ICLR 2022
[4] 3D Infomax improves GNNs for Molecular Property Prediction. ICML 2022
[5] GEOM, energy-annotated molecular conformations for property prediction and molecular generation. Scientific Data 2022
好了,关于KDD 2023 | 第四范式开发用于分子性质预测的生成式3D预训练模型就讲到这。
版权及免责声明:凡本网所属版权作品,转载时须获得授权并注明来源“科技金融网”,违者本网将保留追究其相关法律责任的权力。凡转载文章,不代表本网观点和立场,如有侵权,请联系我们删除。
相关文章
- “铜陵”化工厂爆炸?两名造谣者被查处
- “爷爷”爷爷的石榴树
- “陈老”高山仰止,景行行止 ——读春桃老师所著《国医》
- “知网”中国知网用户委员会两名成员公开亮相
- “宜宾”近2万人参加!2023宜宾长江马拉松开跑:埃塞俄比亚选手包揽全马组男女前三名
- “鲁南”鲁南制药集团建厂55周年:“向新向未来”
- “民谣”玉林民谣,从成都走向深圳
- “亿元”爱仕达董事长陈合林做铁锅起家 公司已连续亏损两年多他有啥招术?
- “营收”“酱油一哥”黯然失色!市值蒸发超5000亿,海天味业遭转型阵痛
- “可持续”“京澳25”公益计划启航
- “结构”结核杆菌致病机制获揭示
- “口信”支持外贸稳规模优结构,金融监管总局发文加强出口信保公司地市级分支结构管理
- “分子”《科学》(20231013出版)一周论文导读
- “结构性”终于松口了!
- “不法分子”金融监管部门提示:谨防校园贷款连环套、“AI换脸”等新型诈骗手段
- “北京大学”不法分子冒用黄益平教授名义推荐股票,北大国发院:已报案
- “核糖核酸”我国科学家揭示外源核酸诱导的原核生物短Ago蛋白系统发挥功能的分子机理
- “原子”详解2023年诺贝尔物理学奖
- “核糖核酸”原核短Ago系统在病毒入侵前后有啥变化?研究揭示
- “磷光”科学家揭示高分子无序增强室温磷光新机制









